



Rx PRODUCTS
Committed to improving quality of life through cost-effective medications
CAMBER PHARMACEUTICALS


One Vision. One Mission. One Camber.
Generic Rx, Specialty, OTC
Camber is made up of three groups with one common goal: to be the best generics company in the industry. Camber Pharmaceuticals is rapidly growing and had an industry leading 28 product approvals in 2022. With the support of our parent company, Hetero, we are poised to have a record number of product approvals and launches in 2023.
News and Events

April 9, 2024
Piscataway, NJ, April 9, 2024–Camber Pharmaceuticals is pleased to announce the addition of Pantoprazole Sodium […]
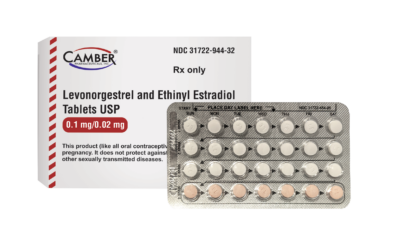
April 4, 2024
Piscataway, NJ, April 4, 2024–Camber Pharmaceuticals is pleased to announce the addition of Levonorgestrel and […]
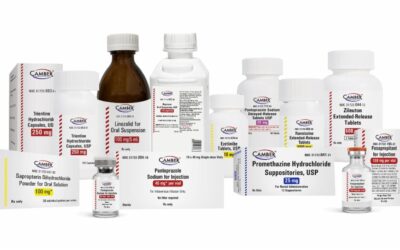
April 3, 2024
Camber Pharmaceuticals is delighted to announce the successful launch of 14 new generic drugs in […]
Camber Specialty Products
Camber Specialty will support the increasing demand for these products by providing hospitals, clinics, government facilities, and other institutional partners more high-quality generic choices.
Camber Consumer Care Over-the-Counter Products
We deliver high quality, over-the-counter medications you can trust. Our mission is to provide a focused and reliable supply of products to the marketplace, creating value for our partners and savings for the consumer.
Learn more

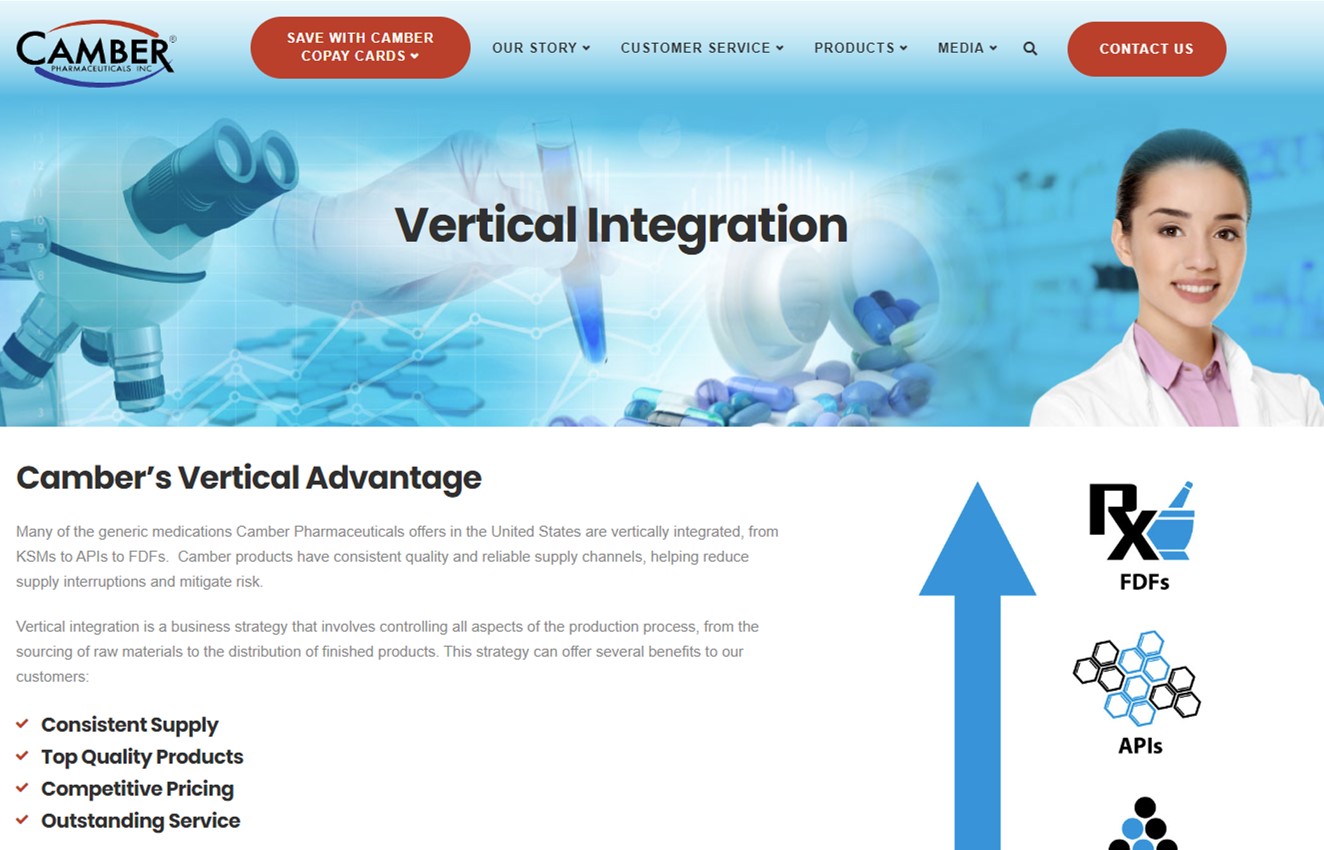
Vertical Advantage
Many of the generic medications Camber Pharmaceuticals offers in the United States are vertically integrated, from KSMs to APIs to FDFs. Camber products have consistent quality and reliable supply channels, helping reduce supply interruptions and mitigate risk.
Business Development
At Camber Pharmaceuticals, our goal is to bring affordable medicines to as many people as possible. As a company, we are continually interested in exploring business partnerships and licensing agreements with strategically aligned companies to support our diverse pipeline of generic products.

